Link: https://www.kaggle.com/c/cafa-5-protein-function-prediction
Problem Type: Multi-label Classification hierarchical label classification
Input:
-
- The fasta files for proteins (aka the amino acids that make it up) AND the corresponding GO terms this protein is related with
- For each GO term, we also learn its aspect:
- Molecular Function (MF)
- Biological Process (BP)
- Cellular Component (CC)
-
- the gene ontology graphs (.obo file):
- basically, for each GO is a “more specific subgraph” of all the aspects above^
- e.g. BP is the root. and it points to three GOs that are more specific
- Then each three has children that are more specific cell functions and so on
- some GOs are leaves.
- biologically, if a cell has a GO, then it also has the GO of all it’s parent GOs up until the root of the tree (either MP, BP, or CC)
- a GO can have multiple parents, so it’s a DAG, not a tree
Output: for each protein, return a set of (GO term ID, probability) for it.
- A GO term is a protein’s function. By returning the set of GO term IDs for the protein, you are saying: “this protein does these set of functions”
- note: the probability is the model saying: “This is the probability that this protein has this function”
- Note: GO means gene ontology
Eval Metric: F-score
- but the precision and recall (to calculate the F1 score) are weighted via the formulas on page 31 of this paper https://ndownloader.figstatic.com/files/7128245
- the weighing function is called information accretion.
Terms deep in the ontology tend to appear less frequently, be harder to predict, and thus their weights are larger (Clark & Radivojac, 2013). This does not always hold true however, as highlighted in the following discussion.
- TODO: understand why their metric implementation was flawed. (there was a discussion post). This will teach you how this metric actually works
Summary
The goal is to figure out “what each protein does” (it’s function)
Note: This is like an oldschool kaggle competition. You submit predictions for the test directly via a tsv (tab separated value) file.
- so competitors know what the test targets are
A good solution uses both the fasta file (getting an LLM to make predictions based on the sequence) and the gene ontology
Important notebooks
- getting started and understanding the competition: https://www.kaggle.com/code/gusthema/cafa-5-protein-function-with-tensorflow
- the majority of the
GO term Ids have BPO(Biological Process Ontology) as their aspect. - there are 40,000 GO term IDs
- but they only plan to make the model predict the top 1500
- they one-hot encode all 1500 into the target array
- it is using the train_embeddings = np.load(‘/kaggle/input/t5embeds/train_embeds.npy’) created using this notebook:
- https://www.kaggle.com/code/sergeifironov/t5embeds-calculation-only-few-samples
- very interesting. They use SeqIO to read the fasta files efficiently (and get the number of sequences efficiently)
- https://www.kaggle.com/code/sergeifironov/t5embeds-calculation-only-few-samples
- I was confused reading the notebook cause I thought the embeddings came from a foundation model, not ones that were trained for the competition labels
- Note: this notebook only uses the fasta file, not the ontology graph to make their predictions
- the majority of the
- explaining information accretion (i.e. how the F-scores are weighed in the evaluation): https://www.kaggle.com/competitions/cafa-5-protein-function-prediction/discussion/405237
- EDA: https://www.kaggle.com/competitions/cafa-5-protein-function-prediction/discussion/418006
- Sequence length is important
- We can see both very short (3 residues) and very long (35375 residues sequences) and they may not be well represented by the LLMs. How to handle them?
- Sequence length is important
Solutions
-
Note: the classes are hierarchical: A leaf class can ONLY exist if all of its parent classes exist.
-
(2nd) graph neural net. conditional probability. gradient boosted trees just hardcore stuff
- https://www.kaggle.com/competitions/cafa-5-protein-function-prediction/discussion/434064
- solution code: https://github.com/btbpanda/CAFA5-protein-function-prediction-2nd-place
- they only did a simple 5-fold CV (nothing else was better)
- Gradient-Boosted Decision Tree improved their models the most (their own Pyboost framework is super fast)
- Alternative modelling approach - predicting the conditional probabilities
- this is very smart. Basically, when training, for each protein, they tell the model: these are the OGs it is.
- but for OGs that are not a parent of a nonzero OG, that value is replaced with NaN, so the model ONLY LEARNS VALID OGs for that protein
- the three values are 0, 1, and NaN.
- 0 since a parent is technically a superclass of the OG, so if the model can’t be as precise and predict the lowest child node, predicting a parent node is still good
- but for OGs that are not a parent of a nonzero OG, that value is replaced with NaN, so the model ONLY LEARNS VALID OGs for that protein
- calculating the conditional probability is also very smart:
p_term_raw = p_term_cond * (1 - (1 - p_parent_0_raw) * (1 - p_parent_1_raw) * ... * (1 - p_parent_N_raw))- and processing the parent nodes first, so the child nodes can use the parent probabilities just makes sense. very cool
- tbh I’m not sure what this is. gotta look at the code
- in which scenarios is the target 0 or 1?
- ik when it’s NaN, but itss not clear.
- how can setting some values to 0 or 1 set the conditiona probability?
- when is something 0???? should it be nan if it’s not a child? I just don’t understand this part
- This is the entire code
trg = np.zeros((num.shape[0], ont.idxs), dtype=np.float32) np.add.at(trg, (trm_ont['n'].values, trm_ont['id'].values), 1) if args.propagate: propagate_target(trg, ont) # create NaNs from graph for k, node in enumerate(ont.terms_list): adj = node['adj'] if len(adj) > 0: na = np.nonzero(np.nansum(trg[:, adj], axis=1) == 0)[0] assert np.nansum(trg[na, k]) == 0, 'Should be empty' trg[na, k] = np.nan- I’m not sure what trm_ont[‘n’].values means. This code is hard to understand without running
- this is very smart. Basically, when training, for each protein, they tell the model: these are the OGs it is.
- used a Graph Convolutional Network (GCN) for the Node Classification Task (using GCN)
- postprocessing
- clip outputs to be within range
- cutoff_threshold_low = 0.1 # prediction < cutoff_threshold_low will be set to zero (i.e. no need to save to submission file)
- by looking at the ontology graph, for the predicted OG, they need make sure that the assigned probability makes sense
- so they look up all the parents for the OG.
- They need to make sure that the predicted probability is never higher than a probability for a parent
- perhaps they get this probability from the training set if it exists
- doing this check is important because their models don’t take this into account (some aren’t aware of the graph relationships)
- They need to make sure that the predicted probability is never higher than a probability for a parent
- clip outputs to be within range
-
(3rd) Used computationally hypothesized GO labels as features
- https://www.kaggle.com/competitions/cafa-5-protein-function-prediction/discussion/464437
- in addition to the ground truth GO labels, there are other GO labels that were computationally hypothesized (non experimental)
- since these aren’t confirmed, he used them as additional training features, rather than as alternative targets (auxiliary objective)
- This^^ is from: “The evidence codes of UniProt GOA annotations”
- Since the test_x will be experimentally validated in the future, he treated the train/validation/private test split like it was time series data
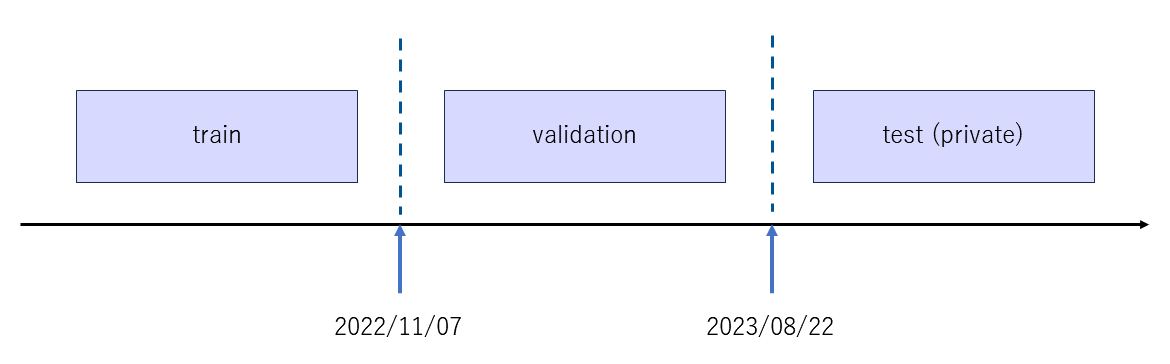
- This feels weird, but I guess it doesn’t matter, cause as long as the validation data is diff form your train it’s good. but it’s weird since you might not be validating your model properly (the metric may not be stable since it’s a bit shifted from your train data)
- but I guess he had to do it this way, cause he knows that in the future, the test (private) data might be collected with more precision, so the drift is expected
-
(4th) extract features using NLP on academic papers
- https://www.kaggle.com/competitions/cafa-5-protein-function-prediction/discussion/433732
- approach
-
- Prot-T5, ESM2, and Ankh Protein Language Model (PLM) embeddings. We carried out no further modifications or finetuning on the output of PLMs, only conversion to float32 to save memory.
-
- A single binary matrix representing species taxonomy for each protein.
-
- extract features using NLP on academic papers: used text information obtained by tf-idf of abstract information from academic papers associated with each protein
-
- from comments:
- concatenating ProtBERT sucked
- dimension reduction for feature generation: However, when I reduced the dimensions of ProtBERT to 3dims using UMAP dimension reduction/tSNE and added it, the score improved
- approach
- https://www.kaggle.com/competitions/cafa-5-protein-function-prediction/discussion/433732
Takeaways
- Using language models (e.g. T5 or ProtBERT) to extract embeddings from the protein sequence is highly recommended