Link: https://www.kaggle.com/competitions/open-problems-single-cell-perturbations
Problem Type: binary classification
Input: cell types and small molecule names
Output: A 18,211 dim vector for each row. These are the probabilities that the molecule (sm_name) will affect each of the 18,211 gene’s expression when applied to this cell_type.
Eval Metric: Mean Rowwise Root Mean Squared Error
Summary
This is basically a regression problem with 2 feature columns and 18211 targets
Note: an estimated 35% of the training data is erroneous: https://www.kaggle.com/code/jalilnourisa/post-eda
- This could affect the validity of techniques used in this competition
Solutions
-
(1st) ChemBERTa
- https://www.kaggle.com/competitions/open-problems-single-cell-perturbations/discussion/459258
- generated features by embedding the description of the cell
- but these text embeddings decreased his score.
- He also tried fine-tuning these text embeddings but it didn’t work
- cross validation: 5fold CV
- ChemBERTa embeddings of the SMILES encodings helped tremendously
- other features he used: the mean, standard deviation, and (25%, 50%, 75%) percentiles per cell type and small molecule
- TODO: what is he aggregating over? A cell type is a class, not a number!
- feature representations
- initial”: ChemBERTa embeddings, 1 hot encoding of cell_type/sm_name pairs, mean, std, percentiles of targets per cell_type and sm_name
- “light”: ChemBERTa embeddings, 1 hot encoding of cell_type/sm_name pairs, mean targets per cell_type and sm_name
- “heavy”: ChemBERTa embeddings, 1 hot encoding of cell_type/sm_name pairs, mean, 25%, 50%, 75% percentiles of targets per cell_type and sm_name
- model selection:
- didn’t work: gradient boosting models, MLP, and 2D CNN
- worked: LSTM, GRU, 1D CNN
- Loss function:
- 0.32MSELoss + 0.24MAELoss + 0.24LogCosh + 0.2BCELoss
- Although BCE is for binary classification, it was used cause it “sends better signals to the models and optimizers when the target values are close to zero”
- e.g.
- BCELoss(0.05, -0.05) = 0.694
- MSELoss(0.05, -0.05) = 0.010 # this loss is much smaller! However, the values are quite far!
- Since most target values are from a Gaussian distribution with mean 0, using BCELoss will more aggressively punish imprecise predictions
- 0.32MSELoss + 0.24MAELoss + 0.24LogCosh + 0.2BCELoss
- removed padding in the ChemBERTa model improved private leaderboard results
- also setting 30% of the input features’ entries to 0 improves the score
- the hypothesis is:
-
- we might not need to know the complete chemical structure of a molecule to know its impact on a cell. OR
-
- there is a biological disorder in the cell, but we still expect it to respond to the drug in the same way
-
- this feels like dropout
- NOTE: significant training data qualities could mean this technique isn’t valid
- the hypothesis is:
-
(2nd) target encoding
- https://www.kaggle.com/competitions/open-problems-single-cell-perturbations/discussion/458738
- Their main source of features were de_train.parquet (which is just the Differential expression value for each gene)
- https://github.com/Eliorkalfon/single_cell_pb/blob/main/utils.py#L67
- then they engineered extra features
- To address high and low bias labels, I utilized target encoding by calculating the mean and standard deviation for each cell type and SM name
- including uncommon columns significantly improved the training of encoding layers for mean and standard deviation feature vectors
- he found targets with significant standard deviation values.
- so he does target encoding (std deviation and mean) on all 18k target variables
- so 36k extra columns
- Note: this is why his code groups by ‘cell_type’, ‘sm_name’ columns. It’s to prepare the data for target encoding
- Notice how he does the appending of these new columns twice:
- once for all the cell_types and once for the sm_name. (so when he trains, he doesn’t miss any)
- so he does target encoding (std deviation and mean) on all 18k target variables
- sampling strategy
- He wanted each model to see many different cells of different types
- so he did a kmeans to cluster each y_value cluster sampling.
- He made sure that each train/test split contained cells from different clusters
- Note: he only included subsets of clusters in the validation set if there were 20+ items in that cluster
- otherwise, all of the items in that cluster would just go into the training set
- He tested various validation percentages between 0.1 - 0.2
- a validation percentage of 0.1 was deemed most effective
- he carefully considered:
- the amount of data needed for a sufficiently large training set
- vs robust validation
- I initially explored utilizing the nn.Embedding layer. However, due to computational constraints on my laptop GPU, I opted for an alternative approach using a linear layer (nn.Linear) to convert sparse features into a dense representation.
- you should probably try feeding sparse features into embedding layers, and dense features into linear layers
- make sure you normalize the dense features (also explained here: https://quoraengineering.quora.com/Unifying-dense-and-sparse-features-for-neural-networks#:~:text=Dense%20features%20incorporate%20information%20from,%2C%20demographics%2C%20keywords%20and%20etc.)
- He wanted each model to see many different cells of different types
- hyperparms:
- weight decay of 1e-4
- model params
- used Huber loss
- used dropout
- used L2 loss
- Implemented gradient norm clipping with a maximum norm of 1.
- Their code on github is REALLY short and easy to read!
-
(3rd) 2-stage prediction. 1: create pseudolables 2: final prediction
-
https://www.kaggle.com/competitions/open-problems-single-cell-perturbations/discussion/458750
-
the sm_name column maps one-to-one with the SMILES column. So he dropped the sm_name column
- he tried using a neural network on the SMILES column but failed.
-
for each of the genes, he plotted the range of possible values and found that the values can be from 4 to 50.
-
he made sure that “Every fold contains one cell type chosen from NK cells, T cells CD4+, T cells CD8+, T regulatory cells”
- only sm_names being in public and private test was involved
- so you don’t train on irrelevant names
- only sm_names being in public and private test was involved
-
has a 2-stage prediction:
-
1st stage - pseudolabel all the test data (255 rows) for more training data: (this is why he’s third!)
- used optuna for all hyperparams:
- dropout%, num neuron in layers, output dim of embedding layer, num epochs, learning rate, batch size, num of dimensions of truncated singular value decomposition
- he used 4-fold CV. but ran each fold twice (prob diff seed / data shuffle)
- the final prediction of this first stage is an ensemble of 7 models
- used optuna for all hyperparams:
-
2nd stage - use train data + pseudolabelled test data
- he used 20 models with diff hyperparams (didn’t mention seeds!)
- more optuna
- Models had high variance, so every model was trained 10 times on all dataset and the median of prediction is taken as a final prediction
- used median over mean!
- he made sure to clip the final predictions based on the min/max of each col (only from the original training data)
-
History of improvements:
- a replacing onehot encoding with an embedding layer
- a replacing MAE loss with MRRMSE loss
- an ensembling of models with mean
- a dimension reduction with truncated singular value decomposition (SVD)
- An example of this is here (not their kernel): https://www.kaggle.com/code/ambrosm/scp-quickstart?scriptVersionId=144293041&cellId=8
- “We denoise the targets by applying a singular value decomposition”
- here is the line of code they actually use fit transform: https://github.com/okon2000/single_cell_perturbations/blob/7e9513972d7cf8ab9c87021bd082712efefff9b9/util.py#L194-L195C6
- an ensembling of models with weighted mean
- using pseudolabeling
- using pseudolabeling and ensembling of 20 models and weighted mean.
What did not work for me:
- a label normalization, standardization
- a chained regression
- a denoising dataset
- a removal of outliers
- an adding noise to labels
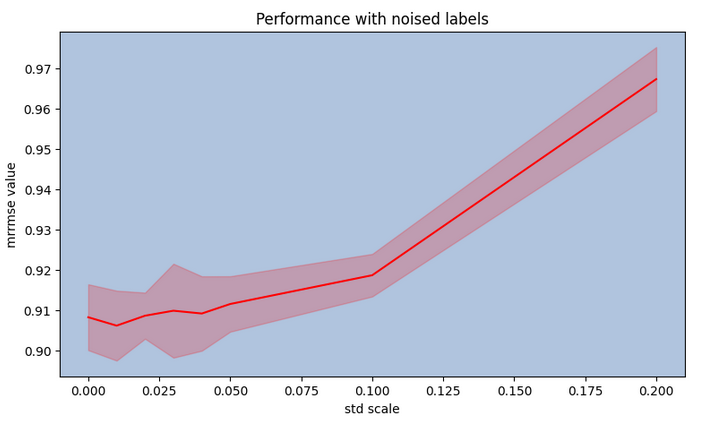
- Adding some noise (0.01 * std) can even improve the model’s performance.
- a training on selected easy / hard to predict columns
- a huber loss.
-
huber loss didn’t work!!! Also outlier removal!
-
-
(4th) good CV + feature engineering
- https://www.kaggle.com/competitions/open-problems-single-cell-perturbations/discussion/460191
- cross validation:
- random k-fold cross-validation wasn’t sufficient. They founds two notebooks with different CV:
- the blue is one test fold of their CV
- https://www.kaggle.com/code/ambrosm/scp-quickstart?scriptVersionId=144293041&cellId=8
- https://www.kaggle.com/code/masato114/scp-quickstart-another-cv-strategy/notebook ^ffdb64
- They went with the second CV (was very consistent between their local CV and LB)
- random k-fold cross-validation wasn’t sufficient. They founds two notebooks with different CV:
- feature engineering for their RAPIDS SVR model
-
- One-hot encoding features of cell_type and sm_name.
-
- use embeddings from ChemBERTa-10M-MTR on SMILES strings
- During the competition, we noticed in the discussion forums that many mentioned the embedding features generated by ChemBERTa-10M-MTR did not yield positive results. In our trials, we found that these features had a negative impact on models like LGBM and CatBoost, but they improved performance in RAPIDS SVR. Therefore, we decided to use RAPIDS SVR to generate pseudo-labels as a basis for subsequent model training.
- When screening features, they only trained and validated the model on the first target: A1BG.
- This tightened their iteration loop to only a few mins
-
- target encoding for cell_type and sm_name
- used [‘mean’, ‘min’, ‘max’, ‘median’, ‘first’, ‘quantile_0.4’]
-
- feature engineering for their Pyboost derived from Alexander Chervov
-
- Pseudo-label features from RAPIDS SVR.
-
- Leave-one-out encoding features for cell_type and sm_name.
- was better than one hot encoding
-
- Embedding features generated by ChemBERTa-10M-MTR for SMILES.
-
- target encoding for [‘mean’, ‘max’].
-
- reduced the dimensionality of 18,211 targets to 45 using TruncatedSVD
- Similarly, we also reduced the dimensions of the above features to the same 45 dimensions
-
- feature engineering for neural net model
- only Leave-one-out encoding for cell_type and sm_name
- other features decreased CV
- also did singular value decomposition (SVD) on the target variables
- They tried converting the SMILES into an image using the rdkit.Chem library
- then training a non-pretrained resnet on it
- but this didn’t help
- ResNet18 was in this model!
- only Leave-one-out encoding for cell_type and sm_name
- 5% of their ensemble went to the 0720 open-source solution
- it uses an “autoencoder method”: https://www.kaggle.com/code/vendekagonlabs/jax-autoencoder-quickstart
- basically, they train an autoencoder on the x-values
- but these x-values are only one-hot-encoded (i.e. they aren’t very good features)
- which is why they only assigned 5% of their ensemble to this
- but these x-values are only one-hot-encoded (i.e. they aren’t very good features)
- I should check to see if they are using the variational autoencoder, or just the standard one in the notebook
- basically, they train an autoencoder on the x-values
- it uses an “autoencoder method”: https://www.kaggle.com/code/vendekagonlabs/jax-autoencoder-quickstart
- Normalization of target data DID NOT WORK


Takeaways
- This was a biological problem that can be abstracted way into important feature selection
- after feature selection, you just needed simple models to get the prediction
-
- using ChemBERTa is important. it gets you very far
-
- target encoding (for mean/std on the cell type / molecule) is important
- the popular public notebook ONLY DID ONE HOT ENCODING: https://www.kaggle.com/code/ambrosm/scp-quickstart?scriptVersionId=144293041&cellId=8
-
- people saw success pseudo labeling the test data and feeding that into round 2 of training
- This way of doing CV is robust: