Link: https://www.kaggle.com/c/stanford-ribonanza-rna-folding
Problem Type: binary classification
Input: RNA sequences (strings)
- you also have other features like
signal_to_noise- (float) Signal/noise value for the profile, defined as mean(measurement value over probed positions)/mean( statistical error in measurement value over probed positions).reactivity_errorcolumns- etc.
Output: the reactivity probability for reactivity_DMS_MaP and reactivity_2A3_MaP for each sequence position id
- https://www.kaggle.com/code/dschettler8845/sr-rna-reactivity-learn-eda-baseline#data_exploration
- i.e. If the sum of the lengths of all 1,343,824 sequences in the test set is 269,796,671, then you should make 2 × 269,796,670 = 539,593,340 predictions.
Eval Metric: MAELoss
Summary
- Ribonanza_bpp_files -
TXTfiles listing position pairs predicted to have non-zero Watson-Crick base pair probabilities by the LinearPartition-EternaFold package. Files are given for train and test sequences, indexed bysequence_id. Note: this package simulates RNA secondary structure ensembles without pseudoknots or other tertiary structure features. It is also limited to Watson-Crick base pairs, even though other kinds of RNA interactions are known to form.
Important notebooks/discussions
- How to check if your model generalizes to long sequences https://www.kaggle.com/competitions/stanford-ribonanza-rna-folding/discussion/444653
- x axis represents position, while y axis represents sequence number
- Since the test set contains longer sequences than the train set, the model doesn’t ‘know’ how to deal with unseen-before positions and does not generalize, i.e., the generated pictures are not similar to the ones posted by @shujun717 above. As for other embeddings, I advise you to experiment and find out what works best
- this picture is also a prediction, not ground truth. So it is possible that your model will do better than theirs.
- so there’s no point in trying to get your model to output a similar image
- sliding window:
- the idea: you train a transformer on sequences of 300 tokens.
- How do you get predictions for sequences of 500 tokens?
- Simple. You just use your 300-token transformer multiple times and take the average of the predictions.
- you slide your 300-token transformer until all 500 tokens were in at least one prediction
- Why they thought it was good for generalization:
- cause the training data was at most 206 long, but the test data is at most 457 long
- so to generalize their models (trained on the shorter training data), they could apply the sliding window on the longer test data examples.
- However, it didn’t work, since it is “wrong in a biological sense” (1st place writeup)
- the idea: you train a transformer on sequences of 300 tokens.
- comments
- About the sliding window, I’m not sure what is the benefit of using it with such short sequences, the main idea behind sliding window is to reduce the complexity from O(n^2) to O(n * w) where w is smaller than n but the RNA sequences of this competition are ⇐ 457, so the normal attention can be computed without much trouble.
-
(1st place): “sliding window seems to be useless”
-
It can be useful in this problem, since in train set Lmax=206, and in test Lmax=457
My guess was if I am to use sliding window attention, my generalization plots would become closer to the ones Shujun shared, but it was not the case, they are still “too sharp”
-
- About the sliding window, I’m not sure what is the benefit of using it with such short sequences, the main idea behind sliding window is to reduce the complexity from O(n^2) to O(n * w) where w is smaller than n but the RNA sequences of this competition are ⇐ 457, so the normal attention can be computed without much trouble.
- Decent EDA:
- https://www.kaggle.com/code/ayushs9020/understanding-the-competition-standford-ribonaza
Solutions
-
(1st) Transformer model with Dynamic positional encoding + CNN for BPPM features
- Squeeze-and-Excitation layer
- To allow better generalization for longer input we implemented Dynamic Positional Bias
- use DBSCAN
- Subsetting data (filtering by different thresholds on SN ratio) resulted in a performance boost for all models. However, this technique was superseded by weight sampling, which proved itself to be more effective.
- by subsetting data, I think they mean: “They only selected training rows based on the signal to noise ratio of each example”
- How did they calculate the signal to noise ratio?
- by weight sampling, I think they mean: “they weighed each training row based on how high it’s signal to noise ratio”
- by subsetting data, I think they mean: “They only selected training rows based on the signal to noise ratio of each example”
- We tried to use data about predicted 3D structure of 100k sequences from the train dataset but gave up on that once we had visually analyzed them:
- probably cause they all looked the same in 3D?
- Using absolute positional embedding leads to unsolvable issues when generalizing upon longer sequences.
- How to solve the positional embedding to generalize to longer sequences
- absolute positional embedding. it doesn’t generalize to longer sequences!
- Try shifting the positional embeddings to the right
- e.g. if our sequence is ends 40 tokens below the context length, we can shift the positional embeddings up to 40 indexes to the right
- so the first token has a positional embedding of 30 (for example)
- e.g. if our sequence is ends 40 tokens below the context length, we can shift the positional embeddings up to 40 indexes to the right
- Rotational positional embedding, unfortunately, doesn’t help the model to generalize on larger lengths.
- ALiBi positional embedding solves the issue with extrapolation but even after keeping it only for a part of heads (as suggested in https://github.com/lucidrains/x-transformers) still behaves worse than dynamic positional bias.
- xpos positional encoding
- Unfortunately, xpos shows rather poor performance on long sequences so we abstained from using this model in the final submission.
- We have also tried shift augmentation and different sequence padding approaches. This didn’t improve our model performance as well.
- remove test data leakage
- 13% of the public test sequences are identical to the ones present in the train dataset (by sequence)
- To avoid selecting a model that is memorizing more of these sequences, we zeroed out the predictions for these sequences
- sometimes we sent non-zeroed out submissions in order to compare our performance to other participants.
- Squeeze-and-Excitation layer
-
(2nd) Squeezeformer layer + BPP Conv2D Attention
- https://www.kaggle.com/competitions/stanford-ribonanza-rna-folding/discussion/460316
- solution code: https://github.com/hoyso48/Stanford---Ribonanza-RNA-Folding-2nd-place-solution
- squeezeformer was the most efficient (compared to newer Conv-Transformer Hybrid architectures)
- showed strong performance early in training and consistently showed faster convergence.
- why use a GRU layer, especially after a transformer layer???
- it just yielded minor improvements. This was probably just intuition
- used ALiBi positional encoding since it’s claimed to generalize better over long sequences than other methods. (it worked better too)
- add signal to attention bias
- this was their model
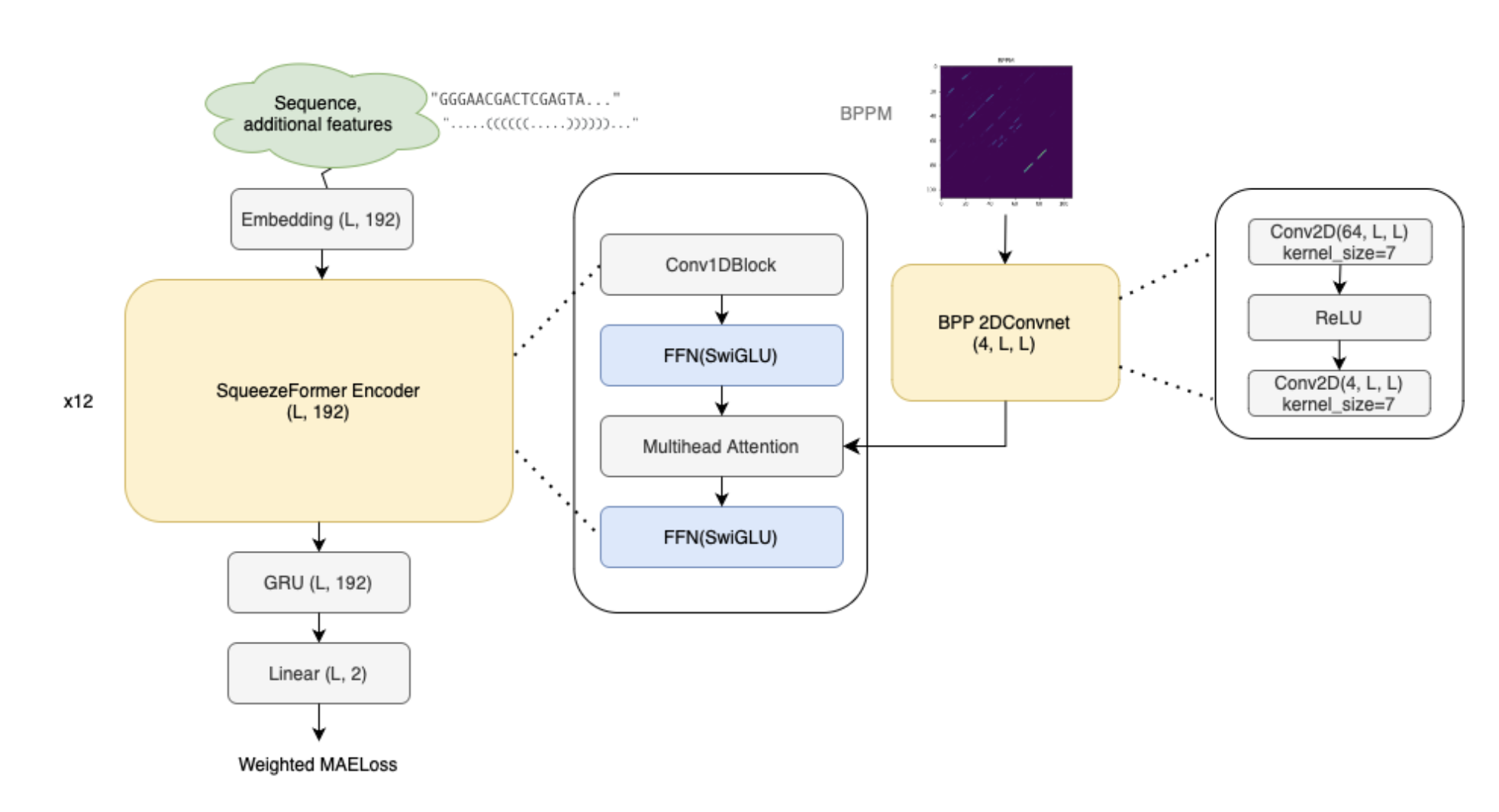
- notice the output of the BPP 2D Convnet layer is fed into the multihead attention layer, specifically, learnable biases
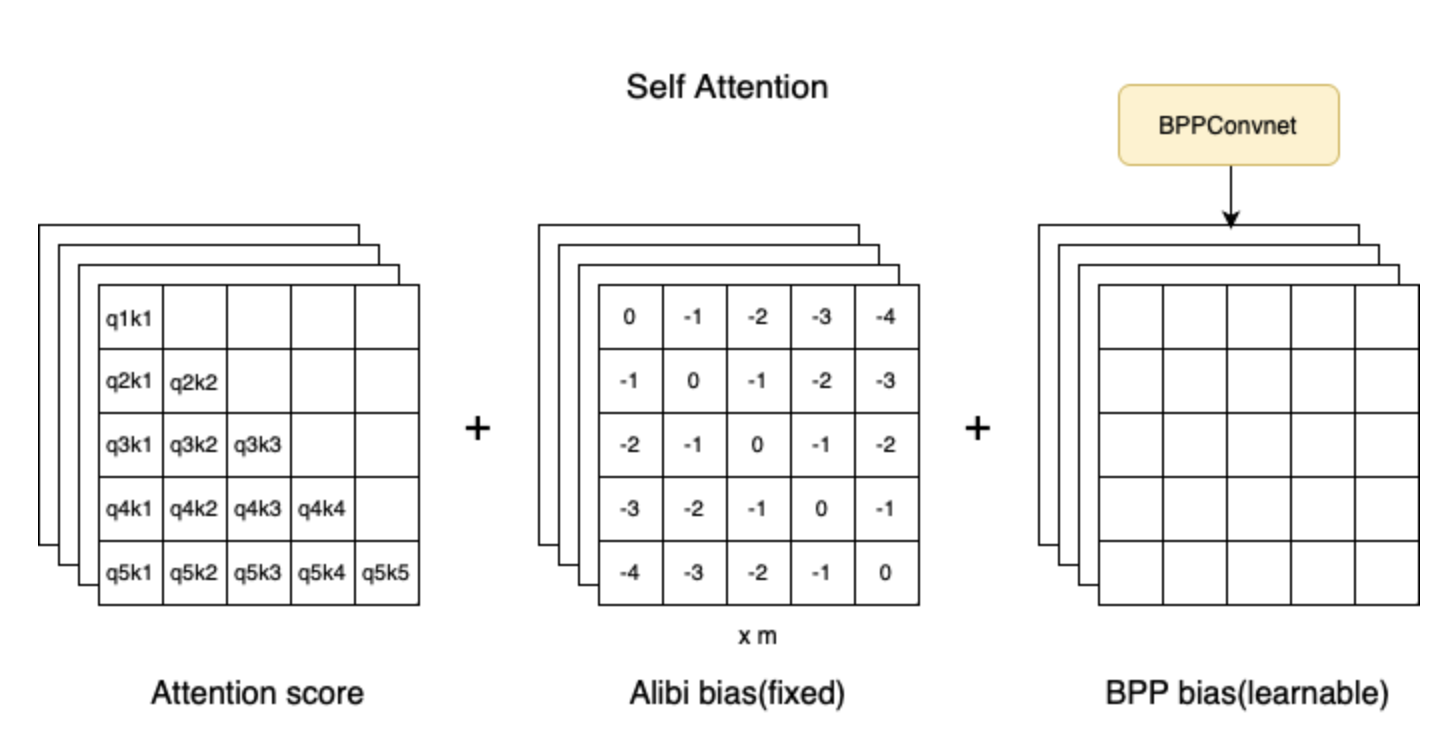
- code
- this was their model
-
(3rd) AlphaFold Style Twin Tower Architecture + Squeezeformer layer
- https://github.com/GosUxD/OpenChemFold
- recycling from alphafold2 wasn’t useful
- this solution VERY inspired by alphafold. that architecture is v complicated and I didn’t look into how it works rn :‘(
Takeaways
- add signal to attention bias using the BPP features (generated from feeding BPP into a conv net) was used by all the top 3 teams